
One of the biggest hidden tax on pharmaceutical innovation today is the burden of global regulatory drag. We work in a world where a single drug dossier can exceed 100,000 pages and must be carefully tailored for dozens of health authorities, including the FDA and EMA, each with its unique and ever-changing rulebook. This has changed the role of the regulatory partner from a strategic business partner to a costly, labour-intensive manual exercise of reconciliation that wastes resources and buries top talent in administrative work.
With this behind us, AI becomes not just a matter of efficiency but a revolution in how we do compliance. By giving the ability to process the regulatory changes with foresight and address the agency question before it is even asked, AI is finally freeing our most skilled experts to concentrate on strategy rather than paperwork. It’s more than just speeding up submissions; it’s re-architecting the relationship between innovation and compliance, transforming a decades-old bottleneck into a cornerstone of competitive advantage.
AI-Driven Document Management & Submission Preparation
Disrupting the way we manage regulatory documents is arguably the most impactful use of AI in pharmaceutical compliance. Manual document creation by teams of specialists has hit its operational limit in a world where a single marketing application could have 100,000+ pages, with interconnected cross-references between modules.
1. Local Prescribed Format Document Generation
Regulatory bodies in the US (FDA), Europe (EMA), Japan (MHLW’s PMDA), as well as other worldwide health authorities, all have stringent filing requirements. These requirements may range from those mandatorily eCTD to other regions like DSHEA compliance in the U.S. To adhere to these requirements, one has to perform manual work that can be time-consuming and error-prone.
AI platforms today automate the formatting of documents with uncanny accuracy that provides several benefits:
Dynamic Reformatting: The submitted document is processed by the AI system, which extracts relevant sections in dynamic reformatting for that region.
For example:
- The underlying AI is designed to conform to FDA eCTD granularity, hyperlinking, and XML backbone rules.
For DSHEA, it generates documents that are specific to DSHEA that include the correct labelling, ingredient listing, and conformity statements.
- Real-time Adaptation: The moment regulators make a change to the submission format, all future submissions will be fully compliant without any intervention.
Multi-Jurisdictional Integration: AI enables submissions to be written for several markets simultaneously, based on a single data set for the submission that has been adapted for each specific regulatory need from market to jurisdiction.
2. Automated Content Selection from Source Documents
Every regulatory submission begins with a stack of source docs — clinical study reports, manufacturing records, nonclinical toxicology studies, and so on. The insight from these unstructured or semi-structured documents is a manual and error-prone process.
AI comes with intelligent content extraction features that make it possible:
- Natural Language Processing: AI systems use NLP to “read” and comprehend unstructured text in clinical study reports, PDFs, and scanned images. They can recognize primary data from secondary operations; pull out critical numbers (e.g., numbers of efficacy endpoints, rates of adverse events), and fill submission templates with little human intervention.
- Contextual Comprehension: AI not only can retrieve the data, but it can also understand its context. For instance, it can help to detect tailored AE profiles in a given treatment population so that only relevant data is incorporated in the submission.
Summary Automation: AI tools can generate structured narratives (e.g., Module 2 summaries for eCTD submissions) from raw data. This saves the regulatory writer from having to cobble together references from hundreds of pages of source.
Intelligent content extraction can speed up submission preparation and improve accuracy, to avoid potential issues with discrepancies and inconsistencies that can result in regulatory delay.
3. Consistency Checking Across Submission Packages
Ensuring a harmonized text while synchronizing thousands of pages in one regulatory submission is an extremely complex task. One discrepancy – For example, a discrepancy in the number of patients from a clinical study in a clinical overview versus a study report — can launch expensive delays and more regulatory questions.
The problem is solved by using AI-powered verification that performs consistency checking.
Semantic analysis: AI applications compare the data and narratives in all parts of a submission package and note any differences in terminology, data points, or conclusions. For example, if a dose value in Module 2.5 does not match a corresponding dose value in Module 5, the system identifies this as an error to be reviewed.
Cross-Referencing validation: Internal cross-references and hyperlinks are validated to ensure they navigate to the appropriate sections and content. Broken or misdirected links are often a problem in a manual submission, and they should be avoided.
Proactive Issue Anticipation: Under the hood, this means using AI to identify questions or objections that health authorities are likely to raise based on historical submissions and past regulatory feedback and flagging parts of the submission that may need better explanation or more data.
Using AI-based consistency checks, regulatory teams may submit their dossiers without the worry of them being sent back for revision or worse, rejected.
Predictive Analytics in Regulatory Strategy
Advanced AI-based predictive analytics is turning regulatory strategy from reactive crisis management to proactive decision making, helping pharmaceutical companies to gain faster approvals and better allocate resources. AI-powered models forecast deadlines by crunching reams of data – from therapeutic classes to regulatory workloads to agency-specific behaviour – permitting companies to synchronize life-sustaining moves, such as manufacturing and commercialization, with projected review dates.
In addition to timelines, these models predict potential health authority questions using natural language processing (NLP) trained on historical agency communications, allowing companies to address shortfalls in clinical and/or CMC data proactively. Optimization of historical data also boosts submission strategies by mining patterns from prior approvals and rejections, including the significance of solid pediatric data for rare diseases or alignment with preferred study designs identified by an agency.
Health authority-specific intelligence is even more granular; AI reads between the lines of regional regulatory differences such as the FDA’s preference for risk-benefit analysis, EMA’s focus on comparative efficacy, or PMDA’s emphasis on local population pharmacokinetics. Customizing submissions with these insights dramatically increases the rate of first-cycle approval and builds good faith with regulators.
Lastly, risk prediction on regulatory pathways provides data-driven decisions on whether to pursue accelerated, conditional or standard approval. AI models run pathway scenarios, measure trade-offs such as earlier market access or more post-approval commitments — putting the sponsor in control to mitigate risk and optimize their investment. This sea change not only speeds patient access to life-saving treatments but also elevates regulatory capabilities as competitive drivers.

Real Time Regulatory Intelligence Systems
A. Automated Global Regulatory Change Tracking
It’s challenging to keep track of over 100 regulatory agencies manually. Today, AI systems consume updates from 500+ sources (agencies, legislative texts, court decisions, draft guidances) in 80+ languages, distilling noise to insight:
Context-Aware Surveillance:
AI not only flags changes — it interprets them. For instance, when Brazil’s ANVISA updated stability testing regulations, the system:
- Auto-translated Portuguese text
- Cross-referenced with existing product formulations
- Tagged affected molecules in the inventory
Notified the stability teams within a few minutes of the publication
Predictive Change Tracking:
NLP reads regulatory meeting minutes/speeches to predict what is coming next (e.g., FDA advisory committee conversations as upcoming changes to oncology endpoint guidance).
B. Impact Analysis: The Path from Regulatory News to Portfolio Valuation Shocks
Static tests do not take business into account. AI quantifies ripple effects:
Portfolio Vulnerability Scoring:
Systems map assets to new requirements using 50+ variables:

Proactive Mitigation Engine: Suggests What You Should Do: “Update Product X Stability Protocol to avoid Brazil Shipment Hold within 45 days” → Link to Template Updates
C. Competitive Intelligence: Unpacking Rivals’ Regulatory Playbooks
Public submissions are treasure troves for strategic foresight. It turns them into chess moves via AI:
- Submission Pattern Analysis:
- Monitors competitors’ approval time lines (e.g., “Rival’s NDA” for similar oncology drug took 247 days vs. industry avg. 300)
Mine’s agency questions to uncover vulnerabilities (“3 FDA queries on rival’s CMC controls → vulnerability in sterile filling”)
Pipeline Gap Detection:
Scans clinical trial registries + approval documents for:
- Pick rival compounds in Phase III
- Backwards build their pathway strategy (i.e., accelerated pathway focus)
Forecast commercial launch well within ±14 days
D. Cross-Disciplinary Collaboration: Shattering the “Regulation Silo”
Real intelligence dies in email alerts. The new systems embed intelligence in the workflow:
Automated Workflow Triggers:
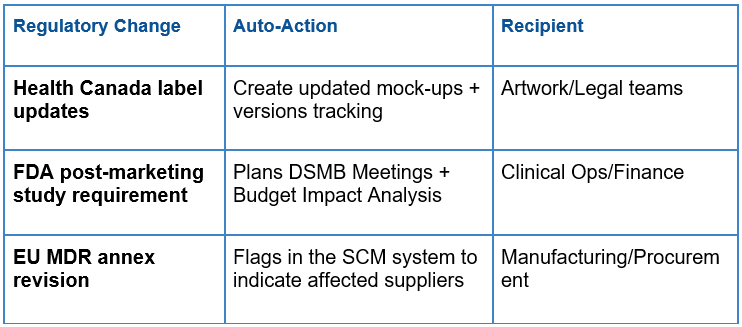
Live – Impact Dashboards:
Real-time visualizations show:
- Portfolio-wide compliance status
- Resource allocation heatmaps
- Revenue-at-risk projections

Compliance Monitoring & Post-Approval Management
Compliance monitoring and post-approval management are entering a transformational new era, underpinned by AI being at the heart of safer and more productive pharmaceutical activities. AI-enabled pharmacovigilance platforms are changing the game for safety monitoring by sifting through large volumes of data (including adverse event reports, electronic medical records, and even social media mentions) to identify safety signals sooner and with more accuracy. Not only do these systems slash the adverse event detection timeline, but they also prioritize the actionable signals, so that the most crucial risks are met head-on.
Automated processing of adverse events also helps in automating pharmacovigilance processes even more by applying NLP to extract, categorise, and code adverse event data – reducing manual reviews and improving accuracy.
In addition to safety, AI systems require ongoing adherence to global regulations. AI models automatically track shifting regulations in real-time and flag new requirements, e.g., post-market safety as well as labelling, and analyze their impact on existing product portfolios to help ensure companies remain one step ahead of compliance issues.
Predictive maintenance of compliance documentation also keeps crucial files, such as clinical reports and manufacturing records, up to date and in line with agency expectations.
Artificial intelligence tools highlight legacy documents, auto-create updates and control versions, thereby decreasing filing lags and regulatory questions. Combined, these offer not just risk reduction, but patient safety, and a more strategic role for regulatory in supporting accelerated entrance to market, operational efficiency, and continuous product success.
Emerging Applications on the Horizon
As new AI applications move beyond automation to become strategic co-pilots, the regulatory environment is set for radical change. Natural language generation (NLG) for regulatory response drafting is advancing from basic templates to intelligent, context-sensitive systems that consider agency queries, historical precedents, and clinical evidence to develop sound scientific arguments in response, decreasing turnaround time and increasing argumentative accuracy.
Generative AI-driven, virtual regulatory affairs assistants are providing instant advice during submission strategy discussions, playing out “what if” scenarios (e.g., EMA’s position on novel endpoints) and calling out compliance blind spots, effectively levelling up expert insights for global organizations.
Blockchain scenarios transform data integrity, providing checksums and audit trails over submitted regulatory documentation. The concept is that anything you sign can be proven later when the FDA or other agency comes to inspect, and data integrity is not arguable.
AI-assisted inspection readiness turns a fire drill into a foresight-driven process: machine learning models dig into historical inspection reports to uncover facility-specific risk patterns, auto-generate corrective action playbooks, and a simulation of how an FDA investigator will question—cutting the preparation time to just a few months at the same time, guaranteeing zero critical findings.
In tandem, these breakthroughs mark the dawn of a new era: from human-driven procedures to AI-coordinated environments, where compliance is no longer about delay and response but the instinctive, predictive virtue – freeing compliance experts to concentrate once again on high-impact strategy while preventing disruptive regulatory aftershocks.

Implementation Challenges & Change Management
The potential of AI in regulatory affairs comes together with four implementation barriers in particular that organisations have to manage strategically.
Data quality and standardization are currently the Achilles’ heel – AI’s predictive potential depends on well-curated and interoperable data, whereas life sciences companies grapple with siloed, non-confirming sources. Successful adopters invest in “data foundations first,” mapping key attributes (CDISC compliant for trials, IDMP standards for products) ahead of any AI rollout.
AI-generated content sees sceptical regulatory acceptance – agencies looking for transparency in AI’s “black box.” Progressive organizations begin using “explainable AI” frameworks by documenting their algorithmic logic and training-data provenance (e.g., embedding audit trails within eCTD submissions) and circulating with the regulators early on through pilot programs such as the FDA’s AI/ML Software Action Plan.
Workforce transformation demands radical reskilling – regulatory professionals need to transition from being document curators to “speaking the language of AI” to validate algorithmic inputs and verify global context. Novartis ‘Regulatory 4.0’ academy scales training in data literacy and oversight of AI, reskilling staff in high-value strategic work.
Balancing automation with expertise entails designing AI to be a copilot, not an autopilot. Human input, however, remains irreplaceable for judgment calls (eg, orphan drug designation negotiations) with leading providers using “three-layer validation”: AI generates responses, experts optimize regulatory strategy, and QA checks templates for agency precedent. The biggest hurdle is not a technologically based one — it’s a cultural one. Firms that get this balance right turn AI from a tactical tool into a regulatory innovation machine, transforming compliance from a cost center into a business accelerator.
Conclusion
Regulators should launch AI pilots targeting key areas (e.g., predictive analytics, automated submissions) and strengthen data chains — good, clean, standardized data is as essential as it ever was. Most importantly, protect the role of humans by transforming teams into AI strategists who interpret outputs, negotiate with agencies, and enforce ethical guardrails.
We need to imbue training with AI literacy to create next-gen capabilities—people who are proficient in all aspects of data science, regulatory tech, and strategic foresight. By 2030, AI will turn regulatory into a value generator from a compliance centre: predicting market shifts 40-60% faster approvals, and driving real-time global compliance – transforming life-saving therapies to market faster and separating agile companies.
Found this article interesting?
1. Follow Dr Andrée Bates LinkedIn Profile Now
Revolutionize your team’s AI solution vendor choice process and unlock unparalleled efficiency and save millions on poor AI vendor choices that are not meeting your needs! Stop wasting precious time sifting through countless vendors and gain instant access to a curated list of top-tier companies, expertly vetted by leading pharma AI experts.
Every year, we rigorously interview thousands of AI companies that tackle pharma challenges head-on. Our comprehensive evaluations cover whether the solution delivers what is needed, their client results, their AI sophistication, cost-benefit ratio, demos, and more. We provide an exclusive, dynamic database, updated weekly, brimming with the best AI vendors for every business unit and challenge. Plus, our cutting-edge AI technology makes searching it by business unit, challenge, vendors or demo videos and information a breeze.
- Discover vendors delivering out-of-the-box AI solutions tailored to your needs.
- Identify the best of the best effortlessly.
Anticipate results with confidence.
Transform your AI strategy with our expertly curated vendors that walk the talk, and stay ahead in the fast-paced world of pharma AI!
Get on the wait list to access this today. Click here.
4. Take our FREE AI for Pharma Assessment
This assessment will score your current leveraging of AI against industry best practice benchmarks, and you’ll receive a report outlining what 4 key areas you can improve on to be successful in transforming your organization or business unit.
Plus receive a free link to our webinar ‘AI in Pharma: Don’t be Left Behind’. Link to assessment here
5. Learn more about AI in Pharma in your own time
We have created an in-depth on-demand training about AI specifically for pharma that translate it into easy understanding of AI and how to apply it in all the different pharma business units — Click here to find out more.